An Overview
Energetics of protein folding. After 1998 I continued to work on the energetics
of protein folding, either by myself or in collaborative work with Franc Avbelj in
Slovenia on the properties of peptide H-bonds. My interest in the stability of the
peptide H-bond (in water) goes back to 1982, when our lab found that, contrary
to the current dogma, a 13-residue peptide fragment of RNAse A is able to form
the (partly stable) N-terminal helix of RNase A in water, in agreement with an
earlier report in 1971 from Werner Klee’s lab. His report was in apparent conflict
with studies of other peptide systems by the labs of Harold Scheraga and Chris
Anfinsen. Our finding led us into an extensive program of analyzing the factors
controlling the stability of peptide helices in water.
My current (2012) interest is in the nature and properties of hydrophobic free
energy, as measured by the energetics of transferring nonpolar solutes between
2 phases, either gas-liquid or liquid-liquid transfers. This interest goes back to
1986, when I found that the temperature dependence of hydrophobic free energy
can be analyzed with a “liquid hydrocarbon model”, based on literature data for
the heat of mixing of liquid hydrocarbons with water.
(Baldwin, RL (1986) PNAS 83:8069-8072).
Ultracentrifuge theory and methods. In the 1950s I developed theory and methods
for making new measurements on proteins and DNA with the analytical ultra-
centrifuge. Still used today is a method for reaching sedimentation equilibrium
rapidly in order to measure protein molecular weights (Van Holde, KE, Baldwin,
RL (1958) J Phys Chem 68:734-743).
DNA physical chemistry. In the early 1960s, our lab developed a physical method
(based on our finding of increased stability of a DNA helix when thymine is
replaced with 5-bromouracil) for determining whether a newly synthesized DNA
strand remains base-paired with its template strand (Inman, RB, Baldwin, RL
(1962) J Mol Biol 5:172-184). Our lab used the method to show that DNA
synthesized in vitro using the Kornberg DNA polymerase remains base-paired
with the template strand (Wake, RG, Baldwin, RL (1962) J Mol Biol 5:201-216).
Later we showed that this is also true of newly synthesized DNA in E. coli
(Shooter, EM, Baldwin, RL (1963) J Mol Biol 7:511-526).
In 1983 our lab developed the cyclization kinetics method of measuring DNA
bending and twisting, in order to understand the free energy changes involved
in bending and twisting DNA in the nucleosome (Shore, D, Baldwin, RL (1983)
J Mol Biol 170:957-981). The results gave the first firm results for the free
energy of DNA twisting.
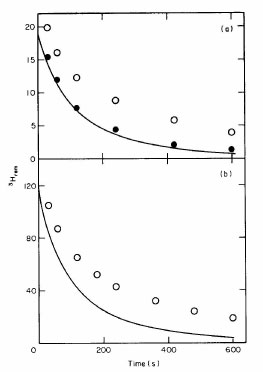
Kinetic intermediates in protein folding. In 1971, by using fast-reaction kinetics,
our lab found the first kinetic intermediates in the reversible unfolding/refolding
reaction of a small protein, RNase A (Tsong, TY, Baldwin, RL, Elson, EL (1971)
PNAS 68:2712-2715). Independently, Charles Tanford’s laboratory found the
same phenomenon in the fast unfolding/refolding reaction of cyt c. The basic
aim of our work was to determine the folding pathway of a protein by characterizing
the structures of on-pathway folding intermediates. This aim has been
controversial for a long time because of arguments over whether or not on-pathway
intermediates exist, or only off-pathway intermediates, and the argument still
goes on today.
In 1973 our lab found there are two different unfolded forms of RNase A, a
fast-folding form and a slow-folding form; the two forms are in (slow) equilibrium
with each other (Garel, JR, Baldwin, RL (1973) PNAS 70:3347-3351). This was
the starting point for work on the role of proline isomerization in protein folding
reactions. In 1975 John Brandts’ lab proposed that the two unfolded forms of
RNase A arise from the slow cis-trans isomerization of proline residues. In 1978
our lab showed that the interconversion reaction of the two unfolded forms of
RNase A is catalyzed by strong acid, and therefore is proline isomerization (Schmid,
FX, Baldwin, RL (1978) PNAS 75:4764-4768).
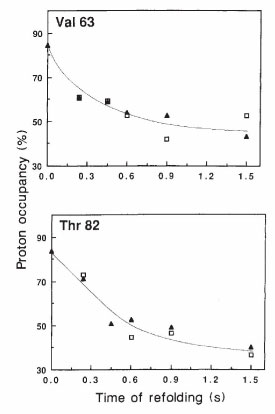
In the late 1980s our lab developed the 1H-2H pulse labeling method for using
NMR to characterize the structures of folding intermediates. I communicated the
method to Heinrich Roder when he was still a PhD student in Kurt Wüthrich’s lab;
Heinrich and Walter Englander later used the method when Heinrich was a postdoc
in Walter’s lab, and our papers were published together in Nature. Our paper was
Udgaonkar, J, Baldwin, RL (1988) Nature 335:694-699. We applied the method in
1990 to determining the structure of the equilibrium molten globule intermediate
of myoglobin, a much simpler system that gave a cleaner result (Hughson, FM,
Wright, PE, Baldwin, RL (1990) Science 249:1544-1548).
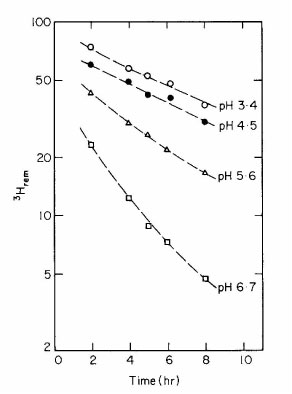
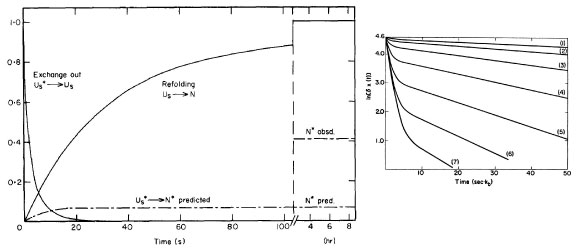
In 1995 our lab found the first evidence for a “dry molten globule” intermediate,
populated in the unfolding of RNase A (Kiefhaber, T, Labhardt, AM, Baldwin, RL
(1995) Nature 375:513-515). This was also the first strong evidence that the dry
molten globule mechanism for protein unfolding, proposed in 1988 by Shakhnovich
and Finkelstein, is correct and our results showed that a populated intermediate is
formed, not a transition-state species as they proposed. The subject remains
controversial today, but new evidence for dry molten globule intermediates in
unfolding has emerged recently.
In 1995, I discussed the emerging arguments over folding pathways between
experimentalists who rely on characterizing folding intermediates (“the classical view”)
and simulators who rely on simulating folding experiments (“the new view”): Baldwin,
RL (1995) J Biomol NMR 5:103-109).
Mechanism of peptide helix formation. In 1982, when our lab confirmed the
1971 report of Brown & Klee that the N-terminal helix of RNase A is present and
partly stable in a 13-residue peptide fragment (“C-peptide”) (Bierzynski, A, Kim,
PS, Baldwin, RL (1982) PNAS 79:2470-2474), it was widely believed that all 20
amino acids have unfavorable helix propensities (<1) and short peptides cannot
show measurable helix formation in water. We found a strong pH dependence
of the stability of the C-peptide helix and suggested the helix is stabilized by a
salt bridge. To find out which residues are essential for helix formation, we
substituted Ala for various C-peptide residues, which led to some interesting
results. When we made a comparative NMR study of the helix present in a longer
(19-residue, “S-peptide”) fragment, we found that the helix stops at Met 13in
S-peptide, just as it does in RNase A, and we concluded that a helix termination
signal is present (Kim, PS, Baldwin, RL (1984) Nature 307:329-334). In 1985,
by varying the charge present on the N-terminal residue of C-peptide, our lab found
that a strong charge-helix dipole interaction affects the stability of the C-peptide
helix (Shoemaker, KR et al. (1985) PNAS 82:2349-2353). This was the first
evidence for a charge-helix dipole interaction in a model peptide system;
similar results were found in a 1982 study from Tatsuo’s Ooi’s lab, but his
results were not interpreted as evidence for a charge-helix dipole interaction.
In 1989 our lab found that alanine is able to form a peptide helix in water by
itself, without the aid of any specific side chain interactions (Marqusee, S,
Robbins, VH, Baldwin, RL (1989) PNAS 86:5286-5290). Later work from our
lab on measuring absolute (not relative) helix propensities showed that Ala is the
only one of the 20 natural amino acids with a helix propensity greater than 1.
Since Ala has only a methyl side chain, this result means that the helix backbone
itself is stable in water. In 1991, in experiments on varying the position of a single
Gly residue in an Ala-based peptide helix, our lab showed that peptide helix
formation is multi-state, not 2-state as commonly believed, and that the Lifson-
Roig theory of the helix-coil transition fits the results quantitatively (Chakrabartty,
A, Schellman, JA, Baldwin, RL (1991) Nature 351:586-588).
I retired and closed my laboratory in 1998.

